

How Profluent’s E1 brings retrieval augmentation to biology
What works in natural language modeling also works in protein engineering!


Why retrieval matters for protein models
Understanding how protein sequence encodes structure and function remains one of the central challenges in the life sciences. Yet most protein language models still treat each sequence as an isolated datapoint. This forces the entire burden of evolutionary context into model parameters, which leads to blind spots in underrepresented families and amplifies the biases of sequence databases. Profluent’s (an Air Street portfolio company) new E1 family demonstrates that this constraint is no longer necessary. Retrieval augmentation, a technique that transformed natural language processing, is now beginning to reshape protein modeling by allowing models to incorporate evolutionary information at the moment of inference rather than storing it all in weights.
E1 is built around this shift. Its design embraces the idea that proteins are best understood in the context of their evolutionary families. Functional motifs, co-evolving residues, and structural constraints emerge most clearly when a sequence is compared to its close relatives. By enabling the model to draw on homologous sequences during training and inference, E1 brings this relational structure directly into computation.
How E1 works
The core architectural idea behind E1 is block-causal multi-sequence attention.
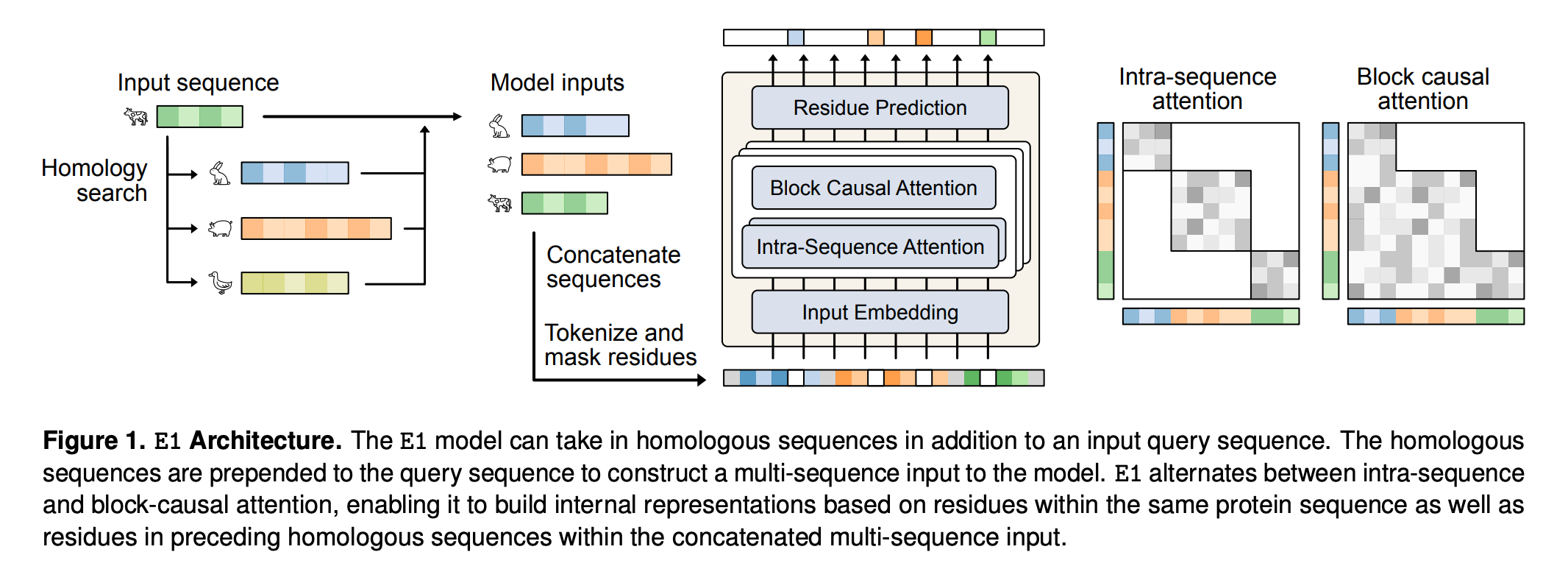
Instead of examining a single protein in isolation, the model receives a query sequence preceded by a set of unaligned homologs. It alternates between layers that attend within each sequence and layers that allow residues to attend across sequences. Because these homologs are unaligned, E1 must implicitly learn how positions correspond across evolutionary space. This mirrors the flexibility that made retrieval so powerful in NLP, where models learn to use external context without relying on rigid structure.
Training further reinforces this capability. Profluent trained E1 on 4 trillion tokens drawn from its proprietary Protein Atlas and UniRef, gradually increasing both the length of multi-sequence instances and the number of homologs included. The result is a family of models at 150M, 300M, and 600M parameters, all released under a permissive license for research and commercial use.
E1 sets the new open-source standard
To understand how this works in practice, consider a protein engineer developing next-generation CRISPR genome editors. Profluent’s earlier model, OpenCRISPR1, showed that language models can learn meaningful relationships among thousands of CRISPR-associated nucleases with minimal supervision. Now imagine an engineer seeking to design a Cas effector variant with improved specificity for a therapeutic context. With E1, they could provide the wild-type nuclease sequence, retrieve its closest evolutionary neighbours from public databases, and ask the model to score how candidate mutations might affect activity or off-target behavior. Because E1 processes the nuclease in the company of its evolutionary relatives, it can highlight conserved positions that tolerate little variation, identify residues whose variation correlates with altered PAM preferences, and surface rare substitutions found only in distant homologs that may expand targeting range.
This kind of enriched reasoning is reflected in benchmark performance. On ProteinGym, E1 establishes a new high-water mark for open-source zero-shot mutation effect prediction. At equivalent parameter scales, all three E1 variants outperform both ESM-2 and ESM C. When homologs are included at inference time, E1 surpasses PoET and MSA Pairformer, reaching an average Spearman correlation of 0.477 and an NDCG at rank ten of 0.788 for the 600M model. The advantages are clearest in protein families with limited multiple sequence alignment depth, where single-sequence models often fail and retrieval provides critical additional signal.
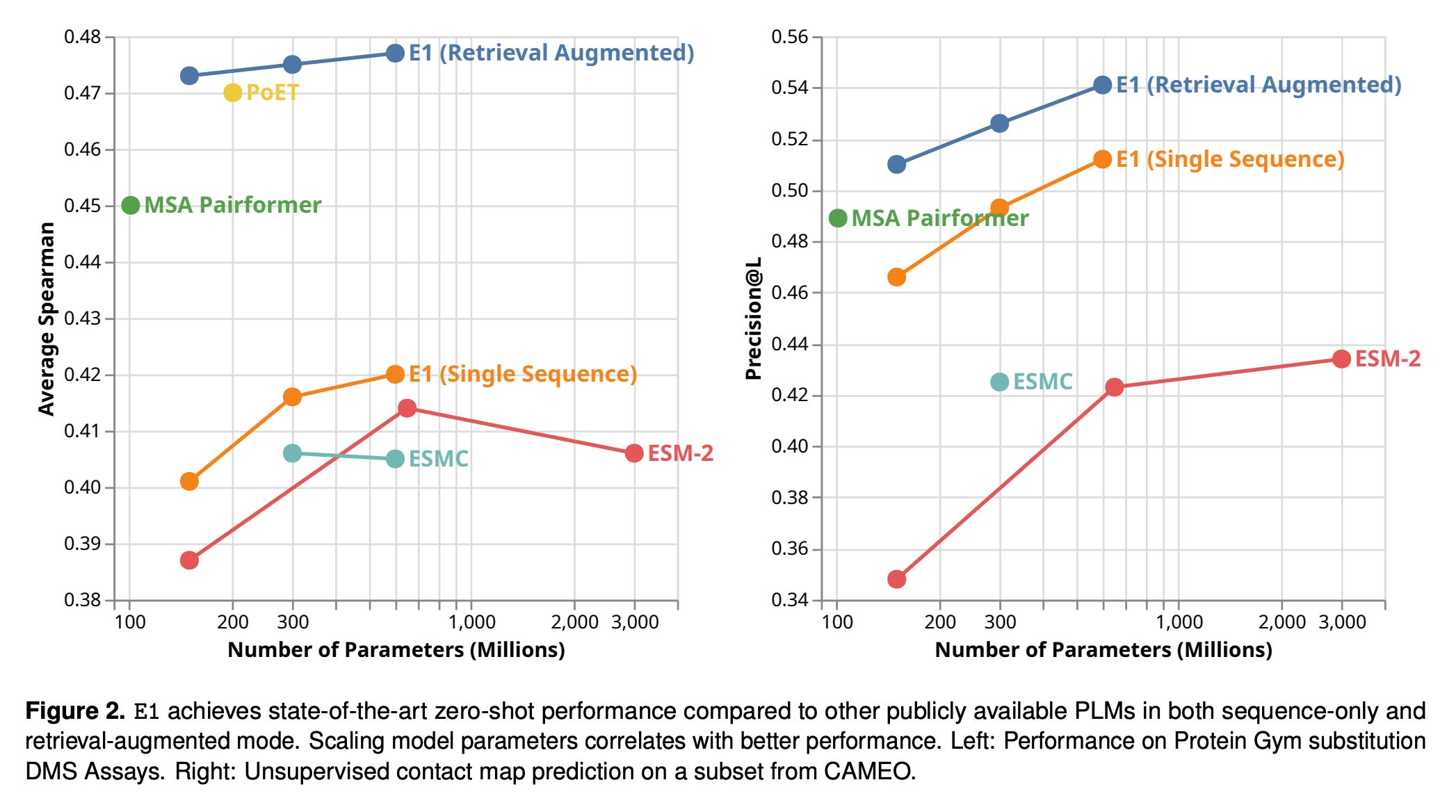
Structural reasoning follows the same trend. Using the Categorical Jacobian method to estimate residue-residue contacts, E1 outperforms the ESM-2 family across every scale. On CAMEO, a continuous blinded benchmark for protein structure prediction, the 600M variant achieves a long-range precision at L of 0.512 in single-sequence mode and 0.541 with retrieval. Examples in the technical report show how retrieval sharpens contact predictions in regions where single-sequence models misinterpret noise as structure.
Taken together, E1 treats evolutionary relationships not as static information distilled during pretraining but as an active force shaping inference. This allows a single model to specialise dynamically for different families without fine-tuning, avoids catastrophic forgetting, and reveals clear scaling behaviour across the released model sizes.
Advancing open protein engineering science
The open release of E1 materially shifts what is possible for research groups and startups working in protein engineering. It offers state-of-the-art performance while remaining small enough to run locally, lowering the computational barrier that often limits exploratory design work. Because the model does not require aligned MSAs, it also simplifies the early phases of design when sequences may be diverse or poorly characterised.
More broadly, E1 shows how ideas first proven in natural language modeling are now regularly diffusing into protein engineering. Retrieval-augmented inference revolutionised document and knowledge retrieval by allowing models to draw on external context at runtime rather than relying entirely on what they had memorised. Profluent has now demonstrated that the same principle applies in biology. By giving a protein model the ability to pull in relevant evolutionary context on demand, E1 represents an important step toward biological foundation models that can flexibly integrate many sources of information.
This cross pollination between AI and biology, where architectural innovations migrate from language to sequence analysis, is likely to guide the next several years of progress. Retrieval may prove to be the missing connective tissue between today’s sequence-only models and future systems that reason jointly over evolutionary, structural, and experimental evidence.

Regarding the topic of the article, its incredibly clever how you connect the power of retrieval augmentation from NLP, which I remember you mentioning before, directly to solving the biases and blind spots in protein modeling, becaus that relational structure is clearly key.